
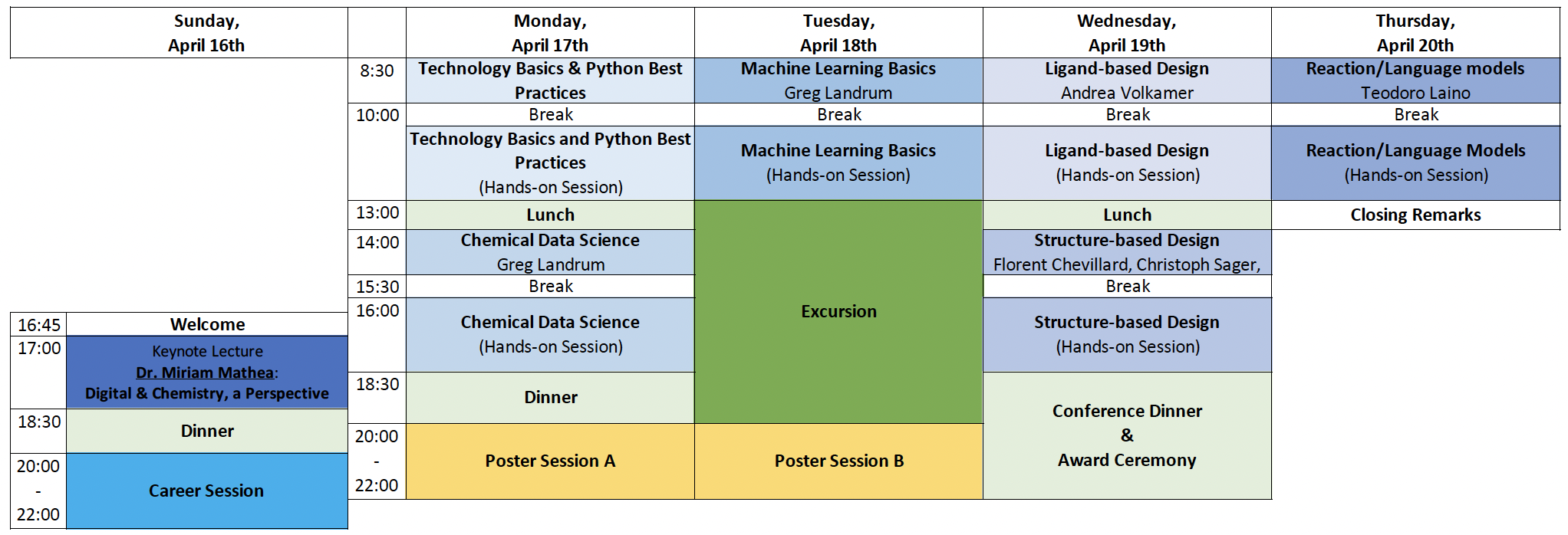
SCS Spring School on Digital Chemistry
«Applied to Drug & Crop Protection Discovery»
16.-20. April 2023, Les Diablerets
SCS Spring School on Digital Chemistry
«Applied to Drug & Crop Protection Discovery»
There is the option to visit Glacier 3000 or chilling in the Wellness Area of the hotel:
https://www.glacier3000.ch/frhttps://www.eurotel-victoria.ch/en/content/multipage-bien-etre-hotel
Python developed in the cheminformatics and machine learning community to the most important programming language. It is easy to get started, but to increase efficiency, reproducibility, and for enabling collaboration, a few aspects need to be considered already at an early stage of code development.
In this first session we introduce and explore the usage of an IDE (Integrated Development Environment), which is a software that was created to make the process easier to write, debug, and to test code in a single environment. We show how to create, activate, and install packages into a Python Virtual Environment. In a subsequent step, we give an overview of key features of Github, and we practice how to create a project, use it for version control and collaborative coding.
In this session we will introduce the most important concepts for working with chemical data using Python. We will look at some of the different file formats in order to understand what they are good (and not so good) for, and then move on to look at some common use cases when working with sets of molecules:- cleaning up a set of molecules in order to get it ready for further analysis
- drawing molecules
- removing duplicates from a set of molecules
- doing substructure searches
- generating fingerprints
We will use the RDKit (https://www.rdkit.org/) and Jupyter notebooks for the hands-on part of the session.
In this session we will cover the fundamentals of machine learning (ML) and its application to chemical problems. The goal is to teach you how to avoid some of the common mistakes people make when applying ML techniques and to give you a foundation you can build on if you want to learn more later. Some of the topics we'll discuss include:
- use cases for machine learning and types of models
- molecular representations: fingerprints and descriptors
- metrics for model quality
- building and testing models
- intro to some key ML algorithms
During the hands-on part of the session we will work with a real dataset using the RDKit (https://www.rdkit.org/), sklearn (https://scikit-learn.org/), and Jupyter notebooks.
In the previous sessions, you have become familiar with programmatic work on molecular data and have acquired some basic knowledge of machine learning. Next, we will explore how we can use this knowledge for computer-aided drug design (CADD).
We will introduce several concepts in the area of ligand-based drug design, scraping compound data from databases (e.g. ChEMBL or PDB), comparing molecules, filtering them (e.g. by ADMET criteria) or detecting meaningful substructures, as well as ligand-based virtual screening, i.e., building your own models for compound activity prediction against a target of choice.
In addition, concepts for structure-based pipelines, including workflows for docking studies, protein-ligand interaction determination and molecular dynamics simulations will be introduced.
The whole session will build on the material of our TeachOpenCADD Plattform (https://github.com/volkamerlab/teachopencadd), in which each topic - theory and code - is provided in an easy to follow jupyter notebook (called talktorial). We invite you to also have a look at the talktorials before the meeting.
Natural language processing models in organic chemistry have emerged as one of the most effective, scalable approaches for capturing human knowledge and modelling chemical processes. Its use in machine learning tasks demonstrated high quality and ease of use in problems such as predicting chemical reactions [1-2], retrosynthetic routes [3], digitizing chemical literature [4], predicting detailed experimental procedures [5], designing new fingerprints [6] and yield predictions [7]. In this session, we will cover the impact of language models in chemistry by highlighting the critical role of NLP architectures in a wide range of digital chemistry tasks and put them into practice with few selected examples.